Lecture 22: Genome Assembly of MRSA Isolates
Learning Objectives
By the end of this lecture, you will be able to:
- Explain two approaches to variant analysis: reference-based calling vs. de novo assembly
- Summarize the epidemiological findings of Hisatsune et al. (2025) on MRSA bloodstream infections in Japan
- Upload paired-end Illumina and Oxford Nanopore data into Galaxy from SRA accessions
- Perform quality control on Illumina reads with
fastp - Run de novo genome assembly using
Flye
1. Where We Are
Our project focuses on understanding mutations in Staphylococcus aureus isolated from Japanese patients with bloodstream infections. In Lecture 20 we ran a variant-calling pipeline—mapping reads to a reference genome and identifying SNPs. In Lecture 21 we used interval operations to intersect those variants with genes of interest.
But S. aureus is highly variable. Each patient can carry a slightly different strain, and a single reference genome cannot capture the full diversity of mobile genetic elements (SCCmec cassettes, prophages, plasmids) that define MRSA lineages. We need a complementary approach.
1.1 Two Approaches to Variant Analysis
There are two fundamentally different ways to characterize genetic variation:
Reference-based variant calling — map reads from each isolate to an official reference (S. aureus NCTC 8325, NC_007795.1) and call SNPs/indels. This is what we did in Lectures 20–21. It is fast and works well for core-genome variants but misses structural variation and accessory genome elements absent from the reference.
De novo assembly — assemble a complete genome for each isolate, then compare assemblies to each other or to a reference. This captures mobile genetic elements, large insertions/deletions, and rearrangements that reference-based calling cannot detect.
We are going to do both. We will re-analyze the data from prior lectures, but first we will create individual assemblies for each isolate.
Within-patient variation is also informative. Reads from a single patient can reveal heterogeneous positions—sites where different alleles coexist within the same infection. This can indicate mixed infections or within-host evolution.
2. The Study: Hisatsune et al. (2025)
Hisatsune et al. [1] extended the Kaku et al. (2022) surveillance to 580 S. aureus bloodstream infection (BSI) isolates collected from 45 Japanese hospitals during 2019–2020. They combined whole-genome sequencing with standardized antimicrobial susceptibility testing and clinical metadata—the first national genomic surveillance of BSI-derived S. aureus integrating clinical data.
2.1 Population Structure
Bayesian hierarchical clustering of the core genome identified 13 distinct sequence clusters (SCs) across three dominant clonal complexes:
- CC1 (26.7%) — primarily ST1, with ST2725 and ST81
- CC8 (21.9%) — almost exclusively ST8
- CC5 (12.6%) — ST5 and ST764
The geographic distribution was uneven: ST1 dominated in eastern Japan while ST8 was more common in the west. SCCmec type IV accounted for 76.5% of total SCCmec, confirming the replacement of type II reported by Kaku et al.
2.2 The High-Risk ST764-SCCmecII Clone
The study’s most striking finding: the ST764-SCCmecII clone showed a 48% 30-day mortality rate—significantly higher than the 22% rate for all other clones combined (p = 0.0088, log-rank test; p = 0.04 after adjusting for age, hazard ratio 1.86). This clone belongs to CC5 and was inferred to have diverged from the New York/Japan clone (ST5-SCCmecII) around 1994 through repeated phage infections carrying superantigen toxin genes and acquisition of antimicrobial resistance genes via mobile genetic elements.
2.3 Antimicrobial Resistance
Comparison of isolates from 2019–2020 with those from 1994–2000 revealed dynamic changes in resistance:
- The median number of antimicrobial resistance genes (ARGs) increased from 3 (1994–2000) to 4 (2019–2020) for MRSA
- ST764-SCCmecII showed the highest median ARG count (5) and high resistance rates to most antibiotics
- Resistance to cefazolin, cefmetazole, gentamicin, clindamycin, and minocycline decreased in MRSA overall
- Quinolone resistance-determining region (QRDR) mutations (GrlA/S80F and GyrA/S84L) increased significantly
2.4 Why Assembly Matters
Many of the key findings—phage integrations, SCCmec cassette structure, plasmid-borne resistance genes—are invisible to short-read reference-based calling alone. The authors performed de novo hybrid assembly (Illumina + Oxford Nanopore) on 59 isolates to resolve these mobile genetic elements. We will replicate this approach on a subset of the same data.
3. Your Assignment: Hybrid Genome Assembly
Each of you has been emailed two SRA accessions from the Hisatsune et al. dataset:
- An Oxford Nanopore (ONT) long-read accession
- An Illumina short-read accession
These are real data from real patients. This is not a pedagogical exercise—this is a genuine reanalysis. If the results are informative, we may pursue publication.
Complete your assemblies by next Tuesday. Contact me immediately if you encounter any issues with Galaxy.
Your analysis consists of three phases:
- Upload data from SRA
- Quality control Illumina reads and filter ONT reads
- Assemble using
Flye
We will examine assembly results next week. Assembly theory will be covered in a dedicated lecture.
4. Flye: A Long-Read Assembler
We will discuss genome assembly algorithms in detail next week. For now, here is a brief overview of the tool you will be using.
Flye [2] is a de novo assembler designed for long, error-prone reads (Oxford Nanopore and PacBio). Unlike short-read assemblers that rely on de Bruijn graphs, Flye builds a repeat graph directly from overlaps between long reads. The key steps:
- Find overlaps — Flye identifies all-vs-all approximate overlaps between reads using a fast disjointig-based approach, tolerating the high error rates typical of ONT data
- Build a repeat graph — overlapping reads are collapsed into a graph where nodes represent unique genomic segments and edges represent adjacencies. Repeats appear as nodes with multiple incoming/outgoing edges.
- Resolve repeats — Flye uses read paths through the graph to untangle repeats, aiming to produce a single contig per chromosome
- Polish — the draft assembly is polished using the original long reads to correct remaining errors. For hybrid assembly, short Illumina reads provide an additional round of polishing (using
Polypolishin the tutorial)
For a bacterial genome like S. aureus (~2.8 Mb), Flye typically produces a single circular contig for the chromosome plus separate contigs for plasmids—far more contiguous than short-read-only assemblies.
Flye has a specific mode for different read types. The tutorial uses “Nanopore corrected” mode. Make sure to select the correct mode when running the tool in Galaxy.
5. Step 1: Uploading Data from SRA
5.1 Download reads from SRA
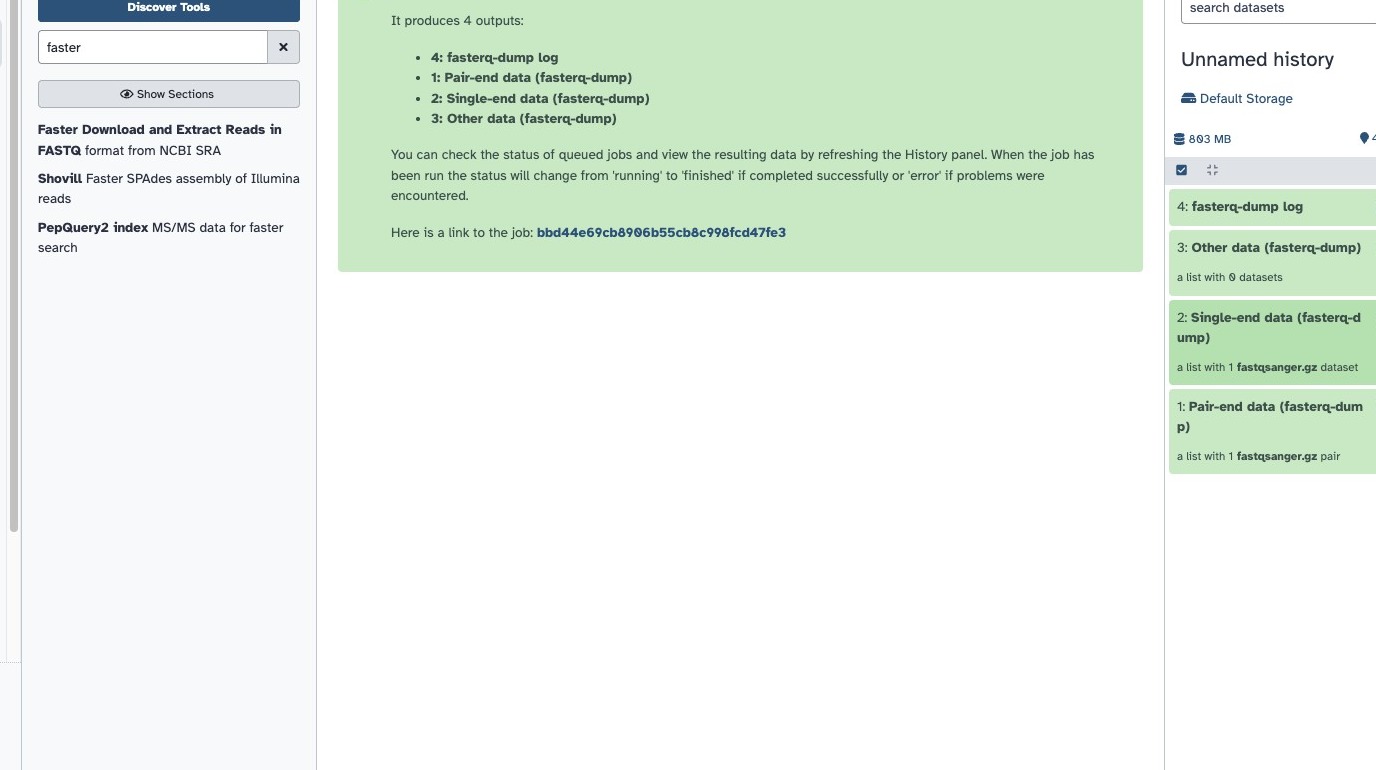
- In Galaxy, find the tool “Faster Download and Extract Reads in FASTQ” (
fasterq-dump)
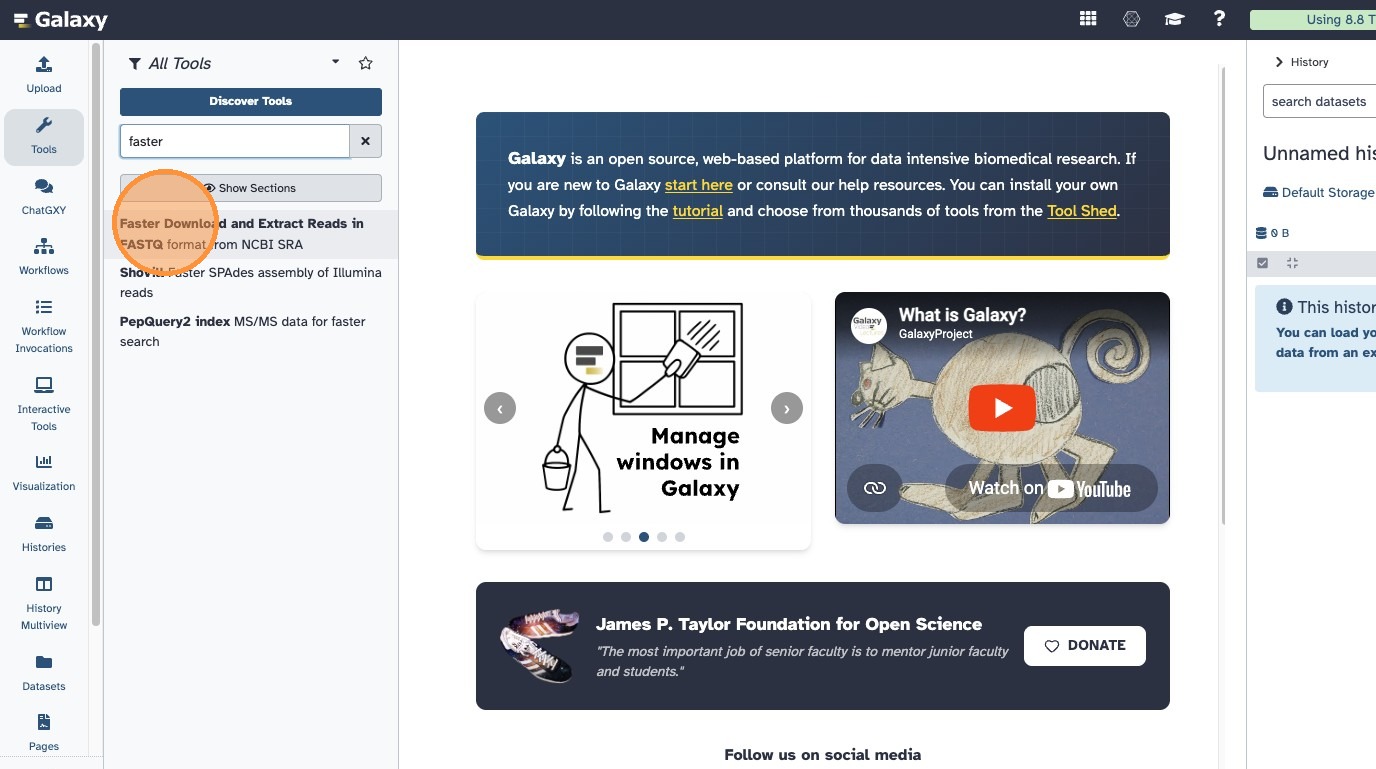
- Enter both accessions you received by email, separated by a comma
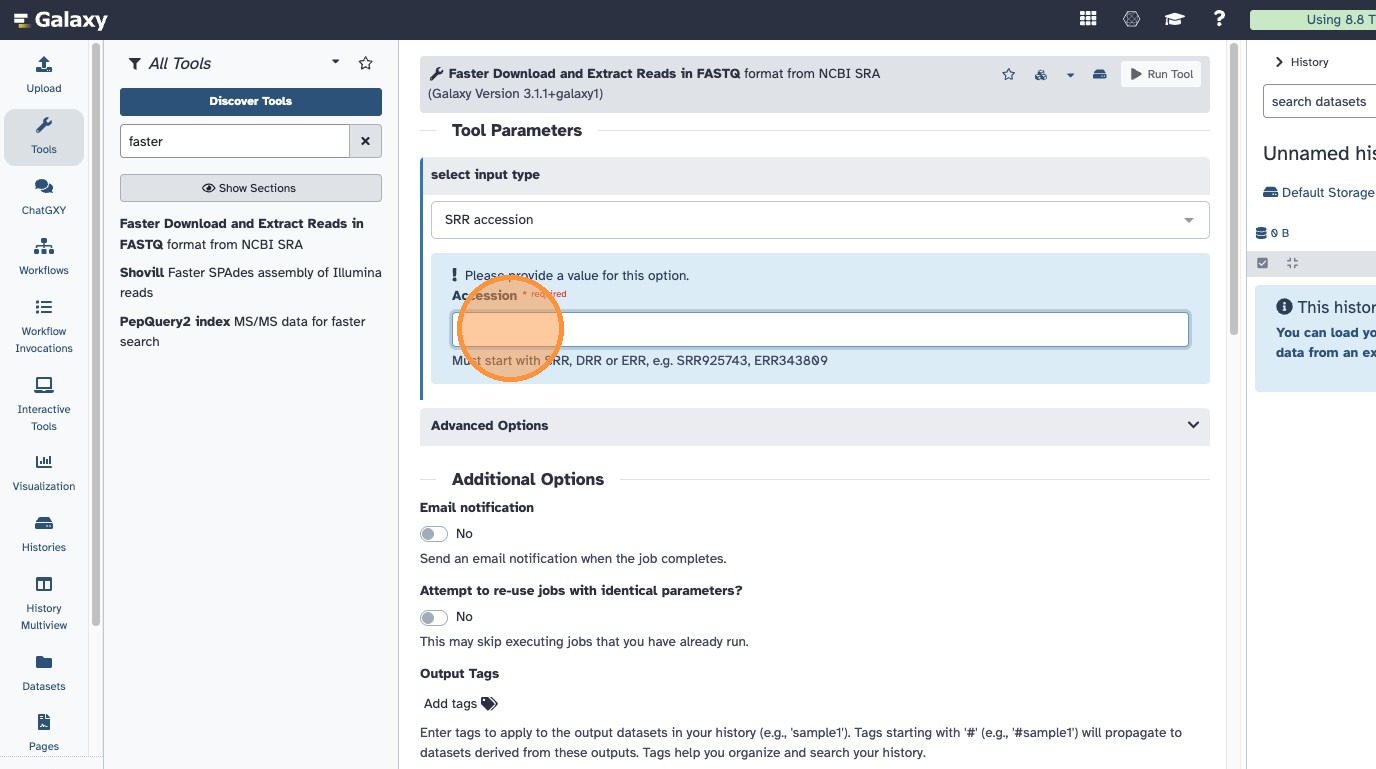
- The input should look like this:
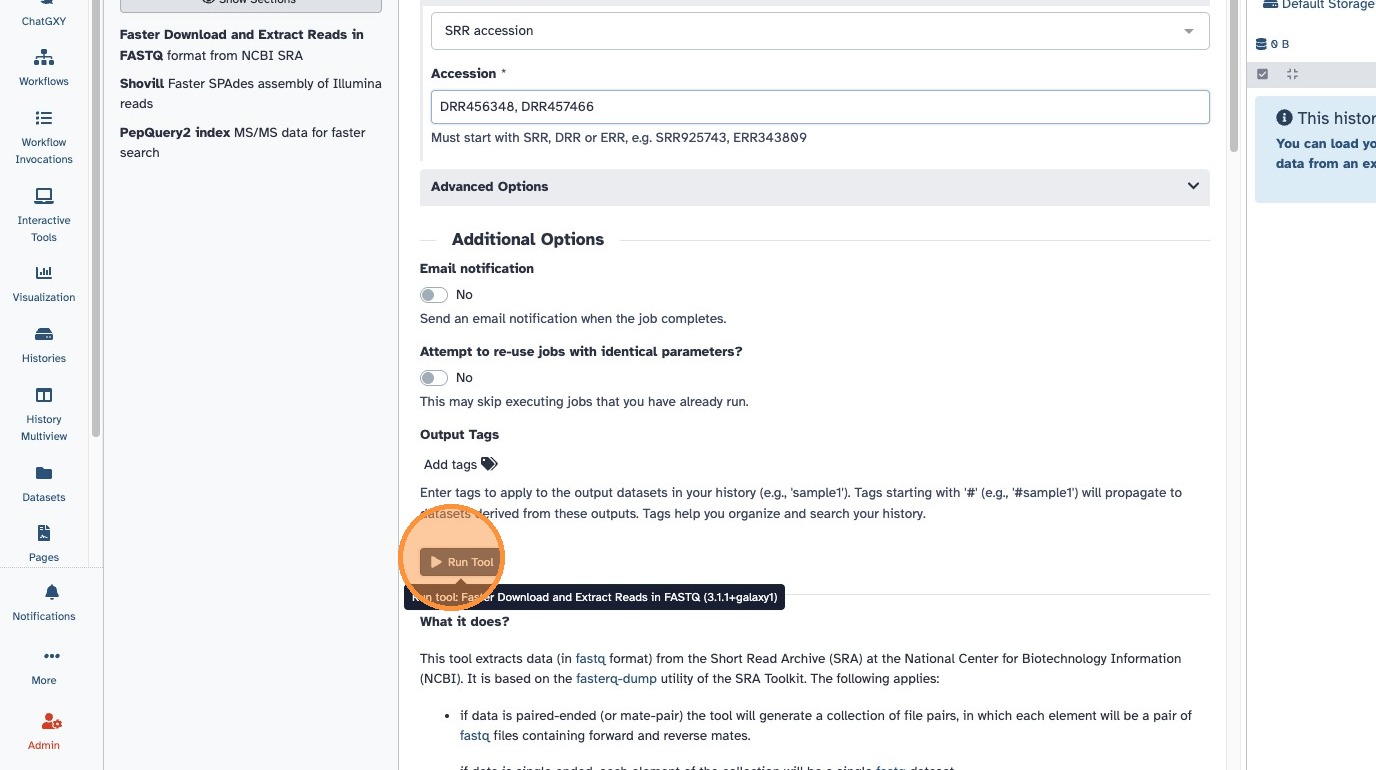
- Click Run and wait for the download to complete
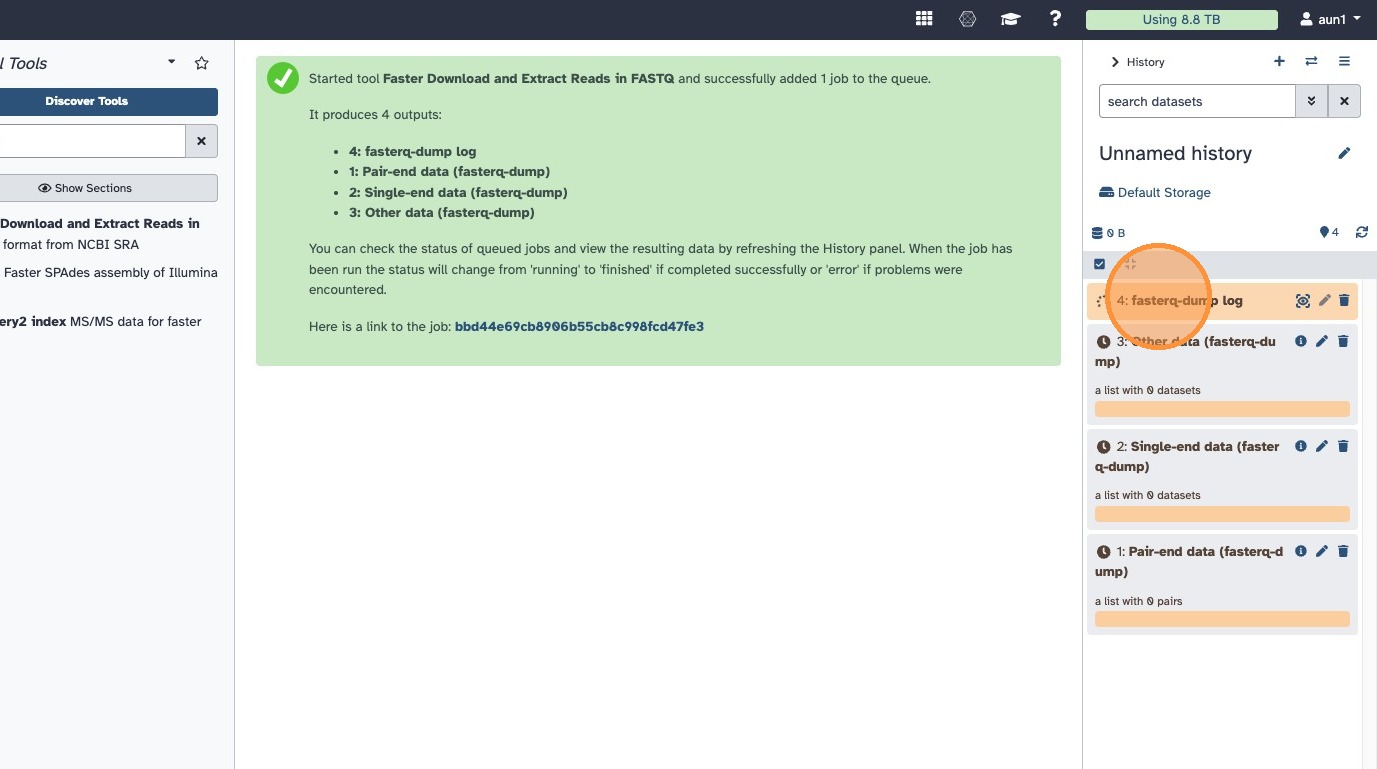
5.2 Identify your output collections
The tool produces two collections:
- Paired-end collection — contains your Illumina data (forward + reverse reads)
- Single-end collection — contains your Oxford Nanopore data
6. Step 2: Follow the Assembly Tutorial
Now that your data is uploaded, follow the Galaxy Training Network tutorial for MRSA Nanopore assembly:
When you reach the section “Hands-on: Choose Your Own Tutorial”, you will see two options. Click “With Illumina MiSeq data” to follow the hybrid assembly path. Do not select the Nanopore-only option—you have both Illumina and ONT data and need the hybrid workflow.
The tutorial walks you through:
- Quality control of Illumina reads with
fastp— trimming adapters and filtering low-quality bases - Filtering ONT reads — retaining high-quality long reads and polishing with Illumina data
- Assembly with Flye — a long-read assembler designed for bacterial genomes; uses repeat graphs to resolve tandem and interspersed repeats. Illumina reads are used for polishing the assembly after construction.
- S. aureus variation requires both reference-based calling and de novo assembly
- Hisatsune et al. (2025) identified ST764-SCCmecII as a high-risk clone with 48% 30-day mortality
- Hybrid assembly (Illumina + ONT) resolves mobile genetic elements invisible to short-read analysis
- Your assignment: assemble your assigned MRSA isolate using
Flyeby next Tuesday
References
- Hisatsune, J. et al. (2025). Staphylococcus aureus ST764-SCCmecII high-risk clone in bloodstream infections revealed through national genomic surveillance integrating clinical data. Nature Communications, 16, 2698.
- Kolmogorov, M. et al. (2019). Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology, 37, 540–546.